This notebook simulates a box of water using the TIP4P-D model and analyzes:
The velocity autocorrelation function (VACF)
The radial distribution function (g(r))
We use OpenMM for simulation and MDTraj + NumPy for analysis.
# Only install conda + OpenMM when running on Google Colab.
# On a local/CI kernel this cell is a no-op.
try:
import google.colab # noqa: F401
_IN_COLAB = True
except ImportError:
_IN_COLAB = False
if _IN_COLAB:
get_ipython().system('pip install -q condacolab')
import condacolab
condacolab.install()
%%capture
!conda install -c conda-forge openmm mdtraj parmedimport openmm
from openmm import *
from openmm.app import *
from openmm.unit import nanometers
import matplotlib.pyplot as plt
import mdtraj as md
import numpy as np
import pandas as pd
import openmmtools
from openmmtools import testsystemsfrom openmmtools import testsystems
#waterbox = testsystems.WaterBox(model='tip4pew', box_edge=3.0*nanometers)
waterbox = testsystems.WaterBox(model='tip3p', box_edge=3.0*nanometers)
system = waterbox.system
topology = waterbox.topology
positions = waterbox.positions
from openmm.app import PDBFile
with open("tip3p_box.pdb", "w") as f:
PDBFile.writeFile(waterbox.topology, waterbox.positions, f)#import nglview as nv
#pdb = md.load_pdb("tip3p_box.pdb")
#view = nv.show_mdtraj(pdb)
#view.add_ball_and_stick('all')
#view.center_view(zoom=True)
#viewSetup simulation in OpenMM¶
nonbondedMethod = PME
nonbondedCutoff = 1.0*nanometers
ewaldErrorTolerance = 0.0005
constraints = HBonds
rigidWater = True
constraintTolerance = 0.000001
hydrogenMass = 1.5*amu
dt = 0.002*picoseconds
temperature = 300*kelvin
friction = 1.0/picosecond
pressure = 1.0*atmospheres
barostatInterval = 25
# Simulation Options
nsteps = int(100000) # run for 1e6 steps = 1ns
dcdReporter = DCDReporter('trajectory.dcd', 100)
dataReporter = StateDataReporter('log.txt', 1000,
totalSteps=steps,
step=True,
speed=True, progress=True,
potentialEnergy=True,
temperature=True,
separator='\t')Run simulation¶
# Prepare the Simulation
system.addForce(MonteCarloBarostat(pressure, temperature, barostatInterval))
integrator = LangevinMiddleIntegrator(temperature, friction, dt)
integrator.setConstraintTolerance(constraintTolerance)
simulation = Simulation(topology, system, integrator)
simulation.context.setPositions(positions)
# Minimize
simulation.minimizeEnergy()
# Simulate
simulation.context.setVelocitiesToTemperature(temperature)
simulation.reporters.append(dcdReporter)
simulation.reporters.append(dataReporter)
simulation.step(nsteps)Analyize Simulation Results¶
# Load trajectory and extract velocities
traj = md.load('trajectory.dcd', top='tip3p_box.pdb')
n_frames = traj.n_frames
n_atoms = traj.n_atomsRDF¶
import numpy as np
import itertools
import matplotlib.pyplot as plt
# Select oxygen atoms
oxygen_indices = traj.topology.select("name O")
# Generate all unique O–O pairs (no self-pairs, no duplicates)
pairs = np.array(list(itertools.combinations(oxygen_indices, 2)))
# Compute radial distribution function
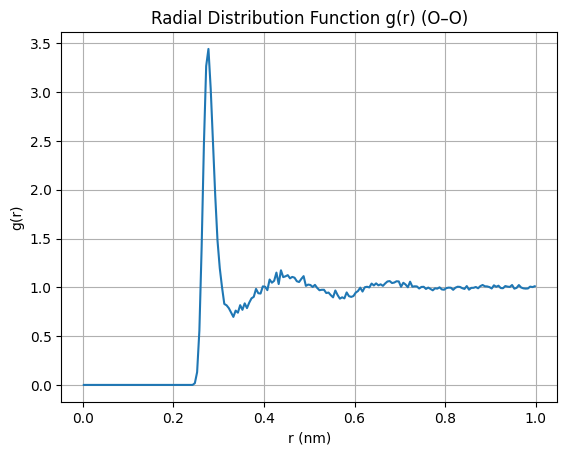
rdf, r = md.compute_rdf(traj, pairs=pairs, r_range=(0.0, 1.0))
# Plot
plt.plot(rdf, r)
plt.title("Radial Distribution Function g(r) (O–O)")
plt.xlabel("r (nm)")
plt.ylabel("g(r)")
plt.grid()
plt.show()
VACF¶
# Compute VACF using NumPy
context = simulation.context
velocities = np.zeros((n_frames, n_atoms, 3))
for i in range(n_frames):
simulation.context.setState(simulation.context.getState(getVelocities=True))
velocities[i] = simulation.context.getState(getVelocities=True).getVelocities(asNumpy=True)._value
vacf = np.zeros(n_frames)
v0 = velocities[0]
# Assume: velocities.shape = (n_frames, n_atoms, 3)
n_frames = velocities.shape[0]
# VACF calculation
vacf = np.zeros(n_frames)
for t in range(n_frames):
vt = velocities[t]
vacf[t] = np.mean(np.sum(v0 * vt, axis=1)) # dot each atom's velocity, then average
# Normalize
vacf /= vacf[0]
# Time in picoseconds (adjust if your frame spacing is not 1 step = 2 fs)
timestep_fs = 2
frame_interval = 10 # if saved every 10 steps
time = np.arange(n_frames) * timestep_fs * frame_interval * 1e-3 # ps
# Plot
plt.plot(time, vacf)
plt.title("Velocity Autocorrelation Function (VACF)")
plt.xlabel("Time (ps)")
plt.ylabel("C_v(t) / C_v(0)")
plt.grid(True)
plt.tight_layout()
plt.show()