Energy of the hydrogen molecule ion#
Using a linear combination of atomic orbitals, it is possible to calculate the best values, in terms of energy, for the coefficients \(c_1\) and \(c_2\). Remember that this linear combination can only provide an approximate solution to the \(H_2^+\) Schr”odinger equation. The variational principle provides a systematic way to calculate the energy when \(R\) (the distance between the nuclei) is fixed:
where \(H_{AA}\), \(H_{AB}\), \(H_{BB}\), \(S_{AA}\), \(S_{AB}\) and \(S_{BB}\) have been used to denote the integrals occurring in variational treatment of the problem. The integrals \(H_{AA}\) and \(H_{BB}\) are called the Coulomb integrals (sometimes generally termed as matrix elements).
This interaction is attractive and therefore its numerical value must be negative. Note that by symmetry \(H_{AA} = H_{BB}\). The integral \(H_{AB}\) is called the resonance integral and also by symmetry \(H_{AB} = H_{BA}\).
Variational solution#
To minimize the energy expectation value with respect to \(c_1\) and \(c_2\), we have to calculate the partial derivatives of energy with respect to these parameters:
Both sides can be differentiated with respect to \(c_1\) to give:
In similar way, differentiation with respect to \(c_2\) gives:
At the minimum energy (for \(c_1\) and \(c_2\)), the partial derivatives must be zero:
In matrix notation this is (a generalized matrix eigenvalue problem):
From linear algebra, we know that a non-trivial solution exists only if:
Energies of bonding and antibonding#
It can be shown that \(H_{AA} = H_{BB} = E_{1s} + J(R)\), where \(E_{1s}\) is the energy of a single hydrogen atom and \(J(R)\) is a function of internuclear distance \(R\):
Furthermore, \(H_{AB} = H_{BA} = E_{1s}S(R) + K(R)\), where \(K(R)\) is also a function of \(R\):
If these expressions are substituted into the previous secular determinant, we get:
This equation has two roots:
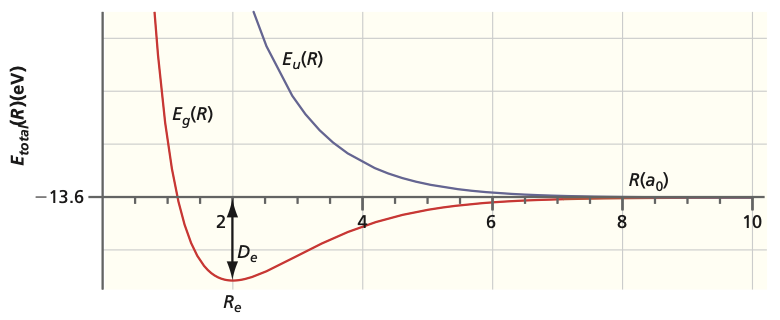
Fig. 94 Energies of bonding and antibonding orbitals#
Since energy is a relative quantity, it can be expressed relative to separated nuclei:
Comparison of MO energies with experiments#
These values can be compared with experimental results. The calculated ground state equilibrium bond length is 132 pm whereas the experimental value is 106 pm. The binding energy is 170 kJ mol\(^{-1}\) whereas the experimental value is 258 kJ mol\(^{-1}\).
The excited state (labeled with \(u\)) leads to repulsive behavior at all bond lengths \(R\) (i.e. antibonding). Because the \(u\) state lies higher in energy than the \(g\) state, the \(u\) state is an excited state of H\(_2^+\).
This calculation can be made more accurate by adding more than two terms to the linear combination. This procedure would also yield more excited state solutions. These would correspond \(u\)/\(g\) combinations of \(2s\), \(2p_x\), \(2p_y\), \(2p_z\) etc. orbitals.
MO diagrams#
It is a common practice to represent the molecular orbitals by molecular orbital (MO) diagrams. The formation of bonding and antibonding orbitals can be visualized as follows:
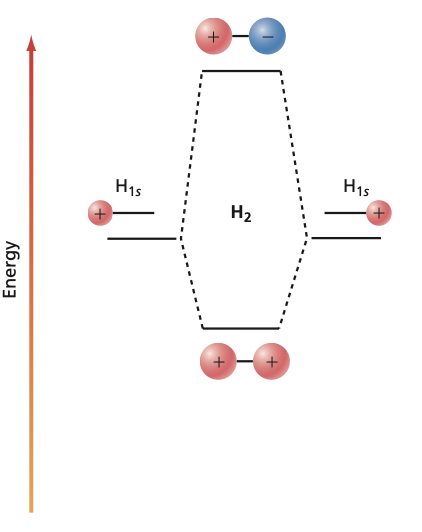
Fig. 95 Molecular Orbital diagram#
\(\sigma\) orbitals When two \(s\) or \(p_z\) orbitals interact, a \(\sigma\) molecular orbital is formed. The notation \(\sigma\) specifies the amount of angular momentum about the molecular axis (for \(\sigma\), \(\lambda = 0\) with \(L_z = \pm\lambda\hbar\)). In many-electron systems, both bonding and antibonding \(\sigma\) orbitals can each hold a maximum of two electrons. Antibonding orbitals are often denoted by *.
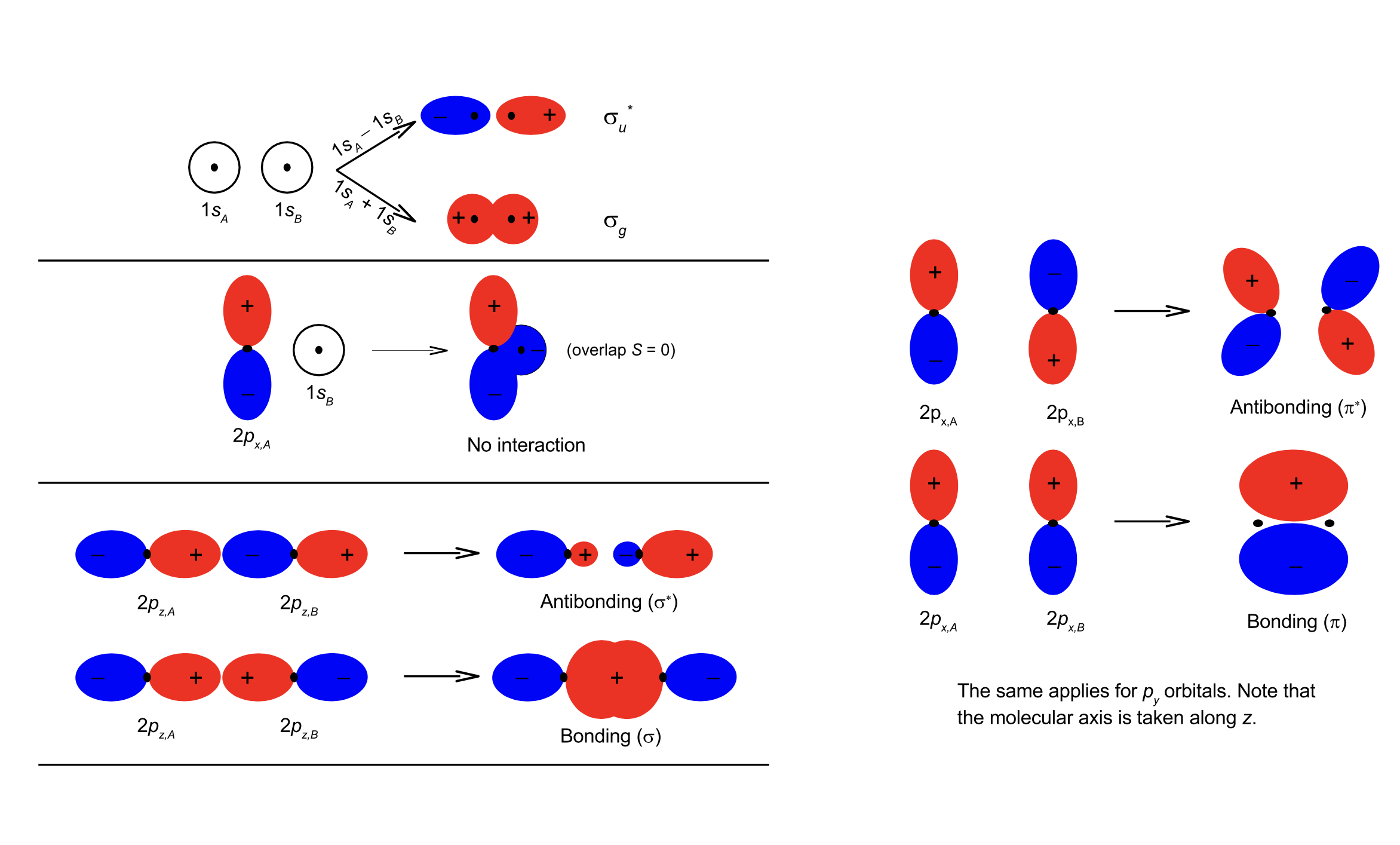
Fig. 96 MOs for homonuclear molecules have distinct symmetry#
\(\pi\) orbitals. When two \(p_{x,y}\) orbitals interact, a \(\pi\) molecular orbital forms. \(\pi\)-orbitals are doubly degenerate: \(\pi_{+1}\) and \(\pi_{-1}\) (or alternatively \(\pi_x\) and \(\pi_y\)), where the \(+1/-1\) refer to the eigenvalue of the \(L_z\) operator (\(\lambda = \pm1\)). In many-electron systems a bonding \(\pi\)-orbital can therefore hold a maximum of 4 electrons (i.e. both \(\pi_{+1}\) and \(\pi_{-1}\) each can hold two electrons). The same holds for the antibonding \(\pi\) orbitals. Note that only the atomic orbitals of the same symmetry mix to form molecular orbitals (for example, \(p_z - p_z\), \(p_x - p_x\) and \(p_y - p_y\)). When atomic \(d\) orbitals mix to form molecular orbitals, \(\sigma (\lambda = 0)\), \(\pi (\lambda = \pm 1)\) and \(\delta (\lambda = \pm 2)\) MOs form.
Excited state energies of \(H_2^+\) resulting from a calculation employing an extended basis set (e.g. more terms in the LCAO) are shown on the left below. The MO energy diagram, which includes the higher energy molecular orbitals, is shown on the right hand side. Note that the energy order of the MOs depends on the molecule.
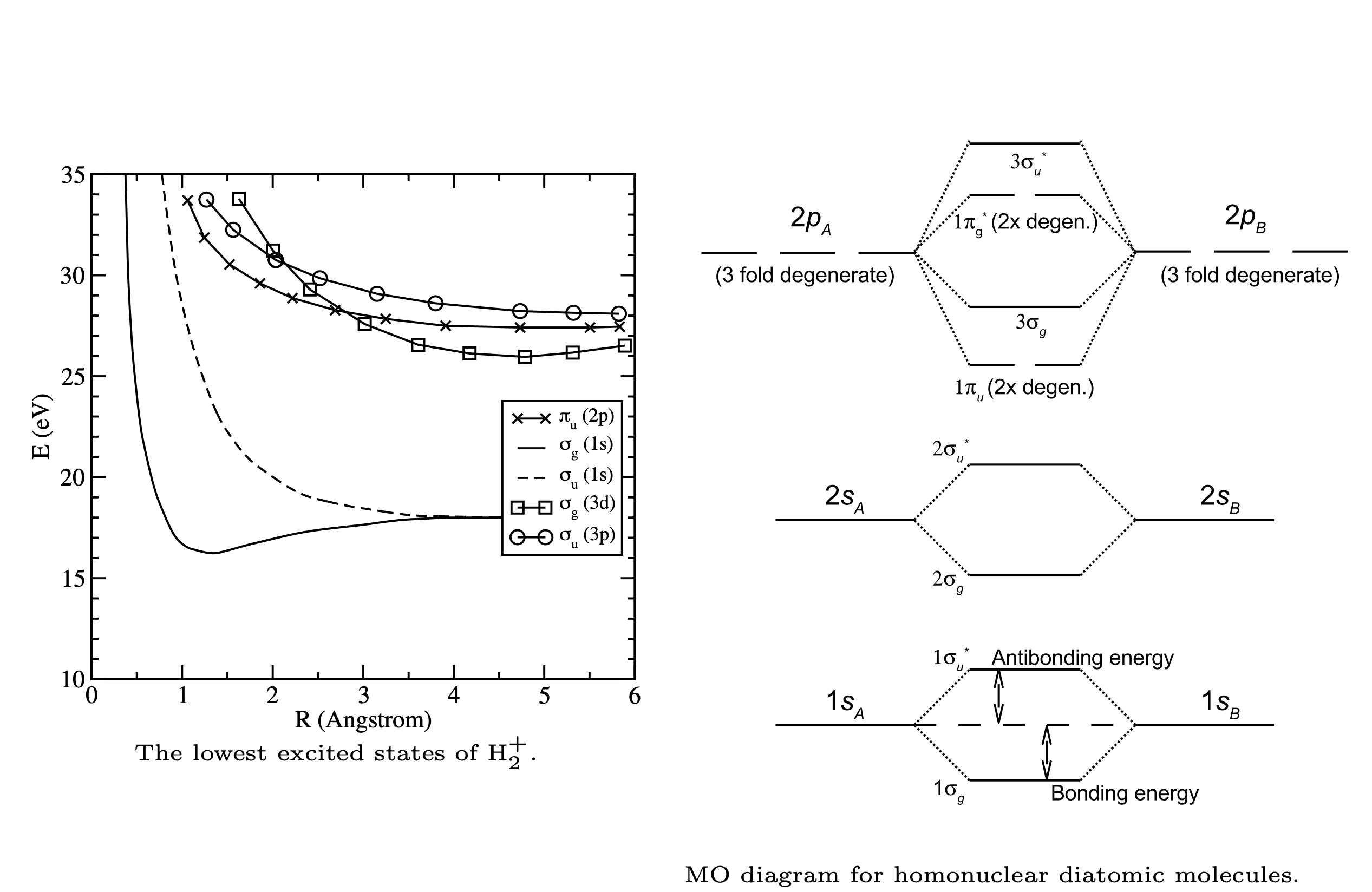
Fig. 97 Mo diagram of homonuclear molecules follows similiar pattern with alternating bonding and anti-bonding MOs.#